


Die neonatale Cholestase, also der Aufstau von Gallenflüssigkeit in den Gallengängen, kann verschiedene Ursachen haben, die in jedem Fall weiter abgeklärt werden müssen. Charakteristisch ist ein cholestatischer Ikterus. Wie ist bei der Abklärung vorzugehen, an welche Differenzialdiagnosen ist zu denken? Und welche Rolle spielt hierbei die Stuhlvisite durch den Pädiater?
Neonatale Cholestase
Die neonatale Cholestase bezeichnet eine Beeinträchtigung der Galleausscheidung, die im Neugeborenenalter beginnt, aber auch weit darüber hinaus andauern kann. Sie stellt keine Krankheitsentität, sondern ein Symptom dar. Die Cholestase kann durch eine Obstruktion oder Fehlanlage der Gallenwege entstehen, ebenso aber auch auf zellulärer Ebene durch eine fehlende Galleausscheidung aus den Hepatozyten. Charakteristisch ist ein cholestatischer Ikterus, der durch eine Erhöhung des konjugierten Bilirubins gekennzeichnet ist. Die neonatale Cholestase betrifft ungefähr eines von 2.500 Neugeborenen [1].
Die neonatale Cholestase kann verschiedene Ursachen haben, die in jedem Fall weiterer Abklärung bedürfen und zumeist auch behandlungsbedürftig sind. Eine große Herausforderung für alle Pädiater stellt die Abgrenzung vom zumeist harmlosen Icterus neonatorum prolongatus dar (vgl. Abb. 1). Ein essenzieller Baustein in der klinischen Diagnostik ist die Stuhlvisite, da viele Eltern die Entfärbung des Stuhlgangs nur schwer in Worte fassen können und nicht als Warnsymptom wahrnehmen.
Die Gallengangatresie ist mit einer Inzidenz von ca. 1:18.000 in Europa zwar eine seltene Erkrankung, aber die häufigste Ursache einer neonatalen Cholestase und die häufigste Indikation für eine Lebertransplantation im Kindesalter [2]. Es handelt sich um eine progressive, fibrotisch entzündlich-destruierende Erkrankung der intra- und extrahepatischen Gallenwege. Man unterscheidet die häufigere nichtsyndromale Form (ca. 80 % der Fälle) von der syndromalen Form, die mit kardiovaskulären Fehlbildungen, z. B. Fallot-Tetralogie oder Vena-cava-inferior-Aplasie, abdominellen Fehlbildungen (z. B. Polysplenie oder Asplenie) und einer Ziliendyskinesie einhergehen kann. Sie tritt auch im Rahmen eines Heterotaxie-Syndroms mit einem häufiger partiellen, seltener totalen Situs inversus auf.
Fallbericht
Ein 5 Wochen alter Säugling mit Ikterus prolongatus
Ein gut 5 Wochen alter weiblicher Säugling wird aufgrund eines Ikterus prolongatus (über den 14. Lebenstag hinaus bestehender Ikterus) in der Sprechstunde der Kinderklinik vorgestellt. Marie (Name geändert) ist das erste Kind gesunder Eltern, nach unauffälliger Schwangerschaft erfolgte die Geburt spontan am Termin. Wegen eines deutlichen Neugeborenenikterus erfolgten am 3., 4. und 5. Lebenstag transkutane Bilirubinbestimmungen, ohne dass eine Phototherapie erforderlich geworden wäre. Im weiteren Verlauf sei der Ikterus zunächst rückläufig gewesen, habe dann farblich etwas changiert und den Orangeton verloren, was insgesamt der Besserung des Befundes zugeschrieben wurde. Bei der U3 im Alter von gut 3 Wochen wurde ein Ikterus prolongatus dokumentiert, optisch als rückläufig wahrgenommen. Bei klinisch unauffälligem, voll gestilltem Neugeborenen und guter Gewichtszunahme wurde auf weitere Diagnostik verzichtet. Der Stuhlgang wurde von der Mutter zu dem Zeitpunkt rückblickend als lehmfarben beschrieben.
 |
Die Ätiologie der häufigeren, nichtsyndromalen Form ist trotz vieler Erklärungsansätze weiterhin ungeklärt. Ätiopathogenetisch werden verschiedene pränatale Virusinfektionen und deren Toxine diskutiert (v. a. Zytomegalievirus (CMV), Epstein-Barr-Virus (EBV), Rota- und Reoviren) [3], ebenso ein autoimmunes Inflammationsgeschehen [4]. Trotz Analyse vieler Kandidatengene erscheint eine genetische Determination unwahrscheinlich, da familiäre Häufungen nur ausnahmsweise auftreten und auch bei monozygoten Zwillingen in der Regel nur eines der Kinder betroffen ist [5].
Die Sonderform der zystischen Gallengangatresie (ca. 5 %) ist häufig bildmorphologisch besonders schwierig zu diagnostizieren und von einer Choledochuszyste zu differenzieren.
Obwohl der zugrundeliegende pathogenetische Prozess vermutlich bereits intrauterin einsetzt, kommen die Kinder in der Regel gesund und eutroph zur Welt. Ein überlagernder physiologischer Neugeborenenikterus mit indirekter Hyperbilirubinämie kann die Diagnose durch einen schleichenden Übergang in den cholestatischen Ikterus wie im hier geschilderten Fall erschweren. Die klinisch apparente Cholestase entwickelt sich häufig erst in den ersten Lebenswochen.
Diagnostik
Klinisch imponieren – auch mit einer bis zu zwei Wochen dauernden Latenz nach der Geburt – der Ikterus, die Stuhlentfärbung und häufig kräftig gelb bis dunkelgelb gefärbter Urin. Eine Stuhlvisite durch den Kinderarzt ist deutlich aussagekräftiger als die Anamnese, da insbesondere Eltern erster Kinder keine Vergleichsmöglichkeiten haben und acholischer Stuhl nicht als solcher erkannt wird. Im Smartphone-Zeitalter ist die Beurteilung der Stuhlfarbe prinzipiell auch durch ein Foto möglich, allerdings muss diesbezüglich die mögliche Farbverfälschung durch die eingebauten digitalen Bildbearbeitungsprogramme bedacht werden.
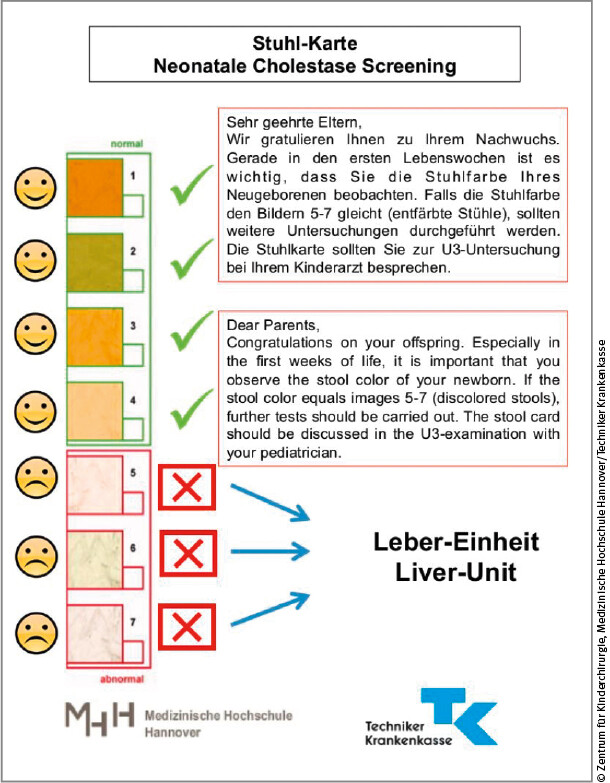
Die in Niedersachsen flächendeckend neu eingeführte Stuhlfarbkarte (vgl. Abb. 2) unterstützt die Eltern bei der Erkennung pathologischer Stuhlfarben.
Bei der syndromalen Form der Gallengangatresie können Herzvitien, Lateralisierungsfehlbildungen und eine Ziliendyskinesie assoziiert sein. Die Kinder wirken im Übrigen in den ersten Lebenswochen gesund und zeigen initial ein gutes Gedeihen, was die Diagnose in vielen Fällen erschwert. Es besteht allerdings bereits nach wenigen Wochen unbehandelter Cholestase ein relevanter Vitamin-K-Mangel mit der Gefahr von Vitamin-K-Mangel-Blutungen. Im Verlauf entwickeln die Kinder aufgrund der mangelhaften Aufnahme kurz- und langkettiger Fettsäuren eine Gedeihstörung. Die Eltern berichten über eine mangelhafte Gewichtszunahme trotz häufigen Trinkens adäquater Milchmengen. Unbehandelt bildet sich über Monate das Vollbild einer biliären Zirrhose mit Aszites, portaler Hypertension und Leberhautzeichen aus.
Laborchemisch finden sich Zeichen der Cholestase mit überwiegend direkter Hyperbilirubinämie – hier ist noch einmal die Bedeutung einer frühzeitigen Bilirubindifferenzierung bei Ikterus prolongatus zu unterstreichen. Zusätzlich sieht man eine Erhöhung von gGT und AP, zudem Zeichen der Leberzellschädigung. Die Lebersyntheseparameter sind in der Regel initial nicht erniedrigt, der durch mangelhafte Resorption der fettlöslichen Vitamine entstehende Vitamin-K-Mangel kann aber frühzeitig zu einer INR-Erhöhung führen.
Sonographisch lässt sich häufig trotz längerer Nüchternphasen keine oder nur eine rudimentäre, kaum gefüllte Gallenblase darstellen – eine sonographisch unauffällig imponierende Gallenblase schließt aber eine Gallengangatresie nicht aus.
Eine hepatobiliäre Sequenzszintigraphie (HBSS) als nuklearmedizinisches Verfahren kann durch Nachweis eines radioaktiv markierten Tracers mit Ausscheidung in das Gallengangsystem der Leber und – bei regulärer Anatomie – in der Folge ins Duodenum eine Gallengangatresie nahezu ausschließen, der fehlende Nachweis einer Galleausscheidung in den Darm kann aber auch bei Differenzialdiagnosen der Gallengangatresie (z. B. alpha1-Antitrypsin-Mangel, progressive familiäre intrahepatische Cholestase) vorliegen [6].
Eine endoskopische retrograde Cholangiopankreatikographie (ERCP) im frühen Säuglingsalter ist technisch extrem anspruchsvoll und die Säuglings-ERCP-Geräte sind nur in wenigen Zentren dauerhaft verfügbar. Bei Darstellung eines regulär angelegten Gallenwegssystems kann so aber eine Gallengangatresie ausgeschlossen werden [7].
Eine Leberbiopsie zeigt bei der Gallengangatresie typischerweise eine intrahepatische Proliferation der kleinen Gallengänge sowie eine lobuläre Cholestase, wichtige Differenzialdiagnosen können histologisch ausgeschlossen werden.
Der Goldstandard ist letztendlich die offene Cholangiographie, bei der operativ eine Sondierung und Kontrastmitteldarstellung der extrahepatischen Gallenwege angestrebt wird. Aufgrund der Invasivität der Methode kommt diese vorwiegend bei hochgradigem Verdacht und in Bereitschaft für eine anschließende operative Hepatoportoenterostomie zum Einsatz.
Differenzialdiagnostik
Die neonatale Cholestase kann verschiedene Ursachen haben, die klinisch in vielen Fällen nur schwierig von der Gallengangatresie zu unterscheiden sind.
Wichtig ist zunächst die Differenzierung, ob eine primär cholestatische Lebererkrankung bei noch erhaltener Leberfunktion vorliegt, oder ob es sich um eine Cholestase im Rahmen eines akuten Leberversagens handelt. Bei primärer Cholestase sind die Kinder initial in der Regel noch in gutem Allgemeinzustand; Gedeihstörung und cholestatische Zirrhose entwickeln sich erst im Verlauf von Wochen bis Monaten. Beim akuten Leberversagen zeigen sich die Kinder in deutlich reduziertem Allgemeinzustand, die Lebersyntheseleistung ist beeinträchtigt und häufig sind auch andere Organsysteme betroffen. Ein niedriger Quick-Wert hilft nicht immer bei der Differenzierung, da dieser auch im Rahmen des Vitamin-K-Mangels bei primärer Cholestase auftritt. Um hier zu differenzieren, kann eine Einzelfaktorenanalyse Vitamin-K-abhängiger und -unabhängiger Gerinnungsfaktoren (z. B. FII und FV) oder eine probatorische intravenöse Vitamin-K-Gabe mit kurzfristiger Verlaufskontrolle erfolgen.
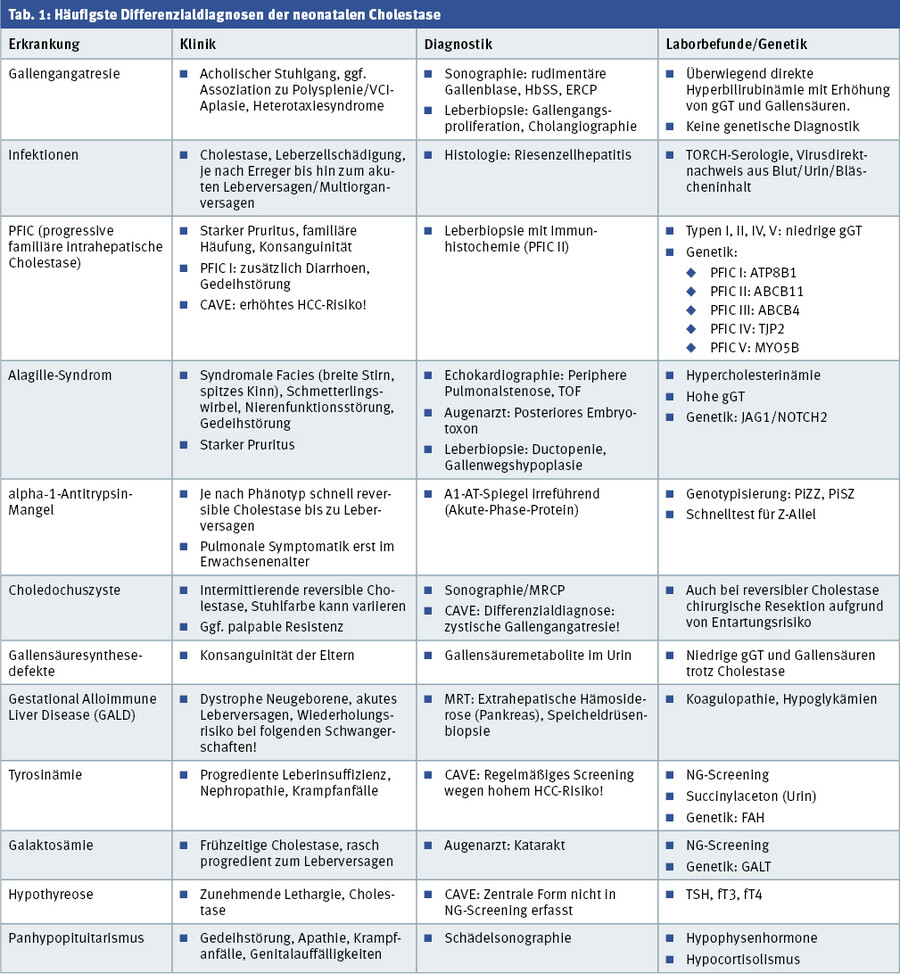
Wichtige Differenzialdiagnosen im Rahmen eines neonatalen Leberversagens mit Cholestase sind eine konnatale Herpes-simplex-Infektion, eine GALD („gestational alloimmune liver disease“, früher „neonatale Hämochromatose“) sowie metabolische Erkrankungen, wie die Tyrosinämie oder Galaktosämie. Diese Erkrankungen manifestieren sich jedoch zumeist früher, führen schnell zur klinischen Verschlechterung und sind bei später auftretender Cholestase und stabilem Allgemeinzustand des Kindes unwahrscheinlich (Tab. 1).
Eine wichtige Differenzialdiagnose ist die neonatale Hepatitis, die sich aufgrund der Unreife des Gallenwegssystems häufig ebenfalls mit einer Cholestase manifestiert. Häufigste Erreger sind CMV, Parvoviren, humanes Herpesvirus 6 (HHV-6) und Enteroviren. Eine neonatale Herpes-simplex-Infektion, oft begleitet durch eine Cholestase, führt in der Regel zu einem akuten Leberversagen innerhalb der ersten Lebenstage, welches häufig in ein Multiorganversagen mündet und eine sehr hohe Mortalität aufweist. Auch eine bakterielle Sepsis kann zu einer Cholestase führen, hier präsentieren sich die Kinder in der Regel aber ebenfalls in deutlich schlechterem Allgemeinzustand.
Die Gruppe der progressiven familiären intrahepatischen Cholestasen (PFIC) kann postnatal ein ähnliches klinisches Bild verursachen, auch hier kann eine Galleausscheidung in der HBSS fehlen. Wichtiger laborchemischer Hinweis ist bei den meisten Formen (PFIC I, II, IV) eine niedrige bis normwertige gGT, da hier durch Transporterdefekte bereits intrahepatisch eine fehlende Ausscheidung von Gallensäuremetaboliten in die Canaliculi der Gallenwege zur Cholestase führt. Die PFIC III (MDR3-Defekt) geht tatsächlich auch mit einer gGT-Erhöhung einher. Diese Erkrankungen können in erster Linie genetisch gesichert werden, die PFIC II lässt sich auch immunhistochemisch in einem Leberbiopsat nachweisen.
Verschiedene Gallensäuresynthese- und -metabolismusdefekte führen ebenfalls zu einer Cholestase und dies möglicherweise bereits im frühen Säuglingsalter. Auch hier ist in vielen Fällen trotz Cholestase die gGT nicht erhöht, zudem zeigen sich die Gallensäuren im Serum, die im Normalfall bei der Cholestase ebenfalls deutlich erhöht sind, in der Regel niedrig oder im Normbereich. Hier gibt eine Analyse der Gallensäuremetabolite aus dem Urin Aufschluss (CAVE: Diese kann durch eine Ursodeoxycholsäure-Therapie verfälscht werden), ebenso die genetische Untersuchung.
Eine neonatale Cholestase mit niedrig normwertigen Serumwerten der gGT und/oder der Gallensäuren macht eine Gallengangatresie als Ursache sehr unwahrscheinlich und sollte immer differenzialdiagnostisch an die Gruppe der PFIC und Gallensäuremetabolismusdefekte denken lassen.
Das Alagille-Syndrom geht mit einer Gallenwegshypoplasie einher und führt auch häufig bereits im Säuglingsalter zu einer Cholestase. Eine auffällige Fazies mit spitzem Kinn und spitzer Nase, eng zusammenstehenden Augen und prominenter Stirn sowie häufige Begleitfehlbildungen (periphere Pulmonalstenose oder andere Vitien, Schmetterlingswirbel, posteriores Embryotoxon) erleichtern die Diagnosestellung in vielen Fällen. Die Erkrankung geht mit einer Gedeihstörung und Kleinwuchs einher, der sich auch nach Lebertransplantation nicht komplett beheben lässt.
Ein alpha-1-Antitrypsin-Mangel kann ebenfalls eine neonatale Cholestase, auch mit Stuhlentfärbung verursachen und ist klinisch mitunter nur schwer von der Gallengangatresie zu unterscheiden. Während ein alpha-1-Antitrypsin-Spiegel im Serum unzuverlässig ist und im Rahmen einer Cholestase generell erniedrigt sein kann, bringt eine Genotypisierung schnelle Gewissheit und sollte vor Durchführung einer intraoperativen Cholangiographie vorliegen [8]. Während die Cholestase häufig reversibel ist, führt sie in seltenen Fällen auch zu einer cholestatischen Leberzirrhose und kann eine Lebertransplantation erforderlich machen.
Des Weiteren kommen endokrine Erkrankungen als Ursache in Betracht. Insbesondere der Hypokortisolismus, häufig im Rahmen eines Panhypopituitarismus, sowie eine Hypothyreose, die in der zentralen Form nicht im Neugeborenenscreening erfasst wird, sollten ausgeschlossen werden [9].
Die häufigsten metabolischen Erkrankungen, die eine neonatale Cholestase und häufig rasch progrediente Leberschädigung verursachen können, sind die Galaktosämie, die Tyrosinämie und in seltenen Fällen auch Harnstoffzyklusdefekte. Auch diese Kinder imponieren krank; die Diagnosestellung erfolgt über das Stoffwechselscreening bzw. anhand einer deutlichen Hyperammonämie. Da hier eine frühzeitige Diagnosestellung und Therapie entscheidend für die Prognose sind, sollten diese Erkrankungen bei entsprechender Klinik und insbesondere bei fehlendem Neugeborenenscreening stets berücksichtigt werden.
Die zystische Fibrose kann ebenfalls mit einer neonatalen Cholestase einhergehen und wird mittlerweile auch zumeist im Neugeborenenscreening durch Bestimmung des immunreaktiven Trypsinogens mit erfasst; dies sollte aber aufgrund der separaten Zustimmungspflichtigkeit stets überprüft werden.
Ein Abklärungspfad der neonatalen Cholestase ist in Abbildung 1 dargestellt.
Therapie der Gallengangatresie
Vor Entwicklung der chirurgischen Behandlungsmethoden sind die Kinder mit Gallengangatresie in der Regel in den ersten zwei Lebensjahren an einer biliären Leberzirrhose verstorben. Im Jahr 1951 entwickelte der japanische Kinderchirurg Morio Kasai das operative Verfahren der Portoenterostomie [10]. Intraoperativ wird zunächst eine offene Cholangiographie zur Darstellung der extrahepatischen Gallenwege angestrebt. Lassen sich diese nicht sondieren und lässt sich somit die Gallengangatresie bestätigen, wird nach Entfernen der extrahepatischen Gallenwegsrudimente die Leberpforte freigelegt und eine seitliche Anastomose zu einer Jejunalschlinge hergestellt. Hierbei wird ein direkter Abfluss aus den in der Leberpforte zusammenlaufenden intrahepatischen Gallenwegen in den Darm geschaffen. Die Jejunalschlinge wird aus der Darmpassage ausgeschaltet und in Y-Technik (Y-Roux-Schlinge) angebracht, um das Risiko von Cholangitiden durch aszendierende Keime zu minimieren.
Der Erfolg im Sinne einer langfristigen Wiederherstellung des Galleflusses ist sehr stark vom Zeitpunkt der Kasai-Operation abhängig; eine zügige Diagnosestellung der Gallengangatresie ist somit entscheidend. Während bei einer Hepatoportoenterostomie in den ersten zwei Lebensmonaten noch bei mehr als 2/3 der Patienten ein Galleabfluss wiederhergestellt werden kann, gelingt das ab einem Lebensalter von über 3 Monaten nur noch bei 25 % der Patienten [6]. Darüber hinaus sind die Ergebnisse natürlich auch von der Expertise des behandelnden Zentrums und der operierenden Kinderchirurgen abhängig. Doch auch wenn die frühzeitig operativ versorgten Kinder bessere Chancen haben, längerfristig mit der eigenen Leber groß werden zu können, stellt die Kasai-Operation tatsächlich nur in der Minderzahl der Fälle eine dauerhaft kurative Therapie dar. Da der entzündlich obliterierende Prozess auch vor den intrahepatischen Gallenwegen nicht Halt macht, wird in der Regel im Laufe des Lebens der Patienten eine Lebertransplantation erforderlich. Im Alter von 15 Jahren haben bereits mehr als zwei Drittel der Patienten eine Lebertransplantation erhalten.
Weiterer Verlauf der im Fallbericht vorgestellten Patientin
Bei Marie zeigten sich keine syndromalen Stigmata. Es ließ sich sonographisch auch nach vierstündiger Nüchternphase keine adäquat gefüllte Gallenblase nachweisen, zudem zeigte sich eine Polysplenie. Laborchemisch ergab sich eine deutliche Cholestase mit über 90 % direktem Bilirubinanteil, erhöhter gGT und erhöhten Gallensäuren sowie Zeichen der Leberzellschädigung mit deutlich erhöhten Transaminasen. Der Quickwert zeigte sich mit 52 % erniedrigt, stieg nach Vitamin-K-Substitution aber wieder auf 87 % an. Hepatotrope Viren ließen sich nicht nachweisen, die Genotypisierung bzgl. eines alpha-1-Antitrypsinmangels ergab einen Wildtyp. Echokardiographisch fanden sich keine Auffälligkeiten, ebensowenig bei der augenärztlichen Untersuchung. Ein Röntgenbild der Wirbelsäule zeigte keine Hinweise für Schmetterlingswirbel. Bei einer nach Priming durch Phenobarbital (Förderung der Bilirubinkonjugation) durchgeführten hepatobiliären Sequenzszintigraphie ließ sich auch in der Spätaufnahme keine Ausscheidung von Galle in den Darm dokumentieren. In einer ERCP ließen sich trotz Papillotomie keine extrahepatischen Gallengänge sondieren. Somit erhärtete sich der Verdacht auf eine Gallengangatresie. Nach fehlender Darstellung der Gallenwege in der intraoperativen Cholangiographie erfolgte am 42. Lebenstag dann die Hepatoportoenterostomie nach Kasai. Postoperativ zeigte Marie einen erfreulichen Verlauf, die Cholestaseparameter waren langsam rückläufig und der Stuhlgang zeigte sich eine Woche nach der Operation wieder gefärbt. Marie erhielt eine Supportivtherapie durch fettlösliche Vitamine, Ursodesoxycholsäure, MCT-haltige Säuglingsnahrung sowie eine antibiotische Cholangitisprophylaxe. Hierunter zeigte sie im Alter von 2 Jahren ein adäquates Gedeihen und eine altersentsprechende Entwicklung. Laborchemisch sehen wir allerdings weiterhin eine mäßige Erhöhung von Transaminasen und gGT sowie sonographisch beginnende Leberumbauzeichen. Marie befindet sich in regelmäßigen Kontrollen in der pädiatrisch-hepatologischen Ambulanz einer Klinik mit Kinderlebertransplantationszentrum, um eine mögliche Verschlechterung der Situation frühzeitig zu erkennen und ggf. eine Listung zur Lebertransplantation in die Wege zu leiten.
Eine wichtige Komplikation, die nachhaltig die Langzeitprognose nach Kasai-Operation beeinträchtigen kann, ist die infektiöse Cholangitis, die durch die direkte Verbindung des Darms zur Leberpforte durch aszendierende Bakterien deutlich häufiger auftritt. Bei Kindern nach Kasai-Portoenterostomie mit Fieber ohne Fokus, insbesondere bei Anstieg der Cholestaseparameter, neu auftretendem Pruritus oder Stuhlentfärbung muss immer an eine Cholangitis gedacht werden.
Als Supportivtherapie ist bei den Kindern auf eine Substitution der fettlöslichen Vitamine zu achten. Solange die Cholestase fortbesteht, sollten die Spiegel von Vitamin A, D und E sowie der Quickwert regelmäßig bestimmt werden; insbesondere Vitamin-K-Mangel-Blutungen sind eine gefürchtete und mit hoher Morbidität verbundene Komplikation der Cholestase. Aber auch der Vitamin-D-Mangel kann zu schweren Komplikationen im Sinne pathologischer Frakturen führen, das Risiko ist auch Monate nach Korrektur der Cholestase noch deutlich erhöht und kann eine zusätzliche Therapie mit Kalzium oder Bisphosphonaten erforderlich machen [11].
Kinder mit chronischer Lebererkrankung haben einen höheren Kalorienbedarf als gesunde Kinder, zudem können aufgrund des fehlenden Galleflusses kurz- und langkettige Fettsäuren kaum resorbiert werden, sodass auf eine Nahrung mit erhöhtem MCT-Fett-Anteil zu achten ist (z. B. Heparon JuniorTM), um eine ausreichende Versorgung mit Fettsäuren zu erreichen. Eine aggressive Ernährungstherapie und somit adäquate Versorgung mit Nährstoffen ist nicht nur wesentliche Voraussetzung für eine adäquate körperliche und geistige Entwicklung, sie verbessert auch signifikant das Langzeitergebnis nach der Lebertransplantation.
Eine Therapie mit Ursodeoxycholsäure sorgt für eine hydrophilere Zusammensetzung der Galle und kann somit den Galleabfluss verbessern. Bei starkem cholestatischem Pruritus, der klassischerweise bei PFIC und Alagille-Syndrom, aber auch bei der Gallengangatresie auftreten kann und die Lebensqualität der Patienten und ihrer Familien massiv einschränkt, gibt es verschiedene Therapieoptionen: Während Antihistaminika keine Besserung der Symptomatik hervorrufen, sind Ursodeoxycholsäure, aber auch Rifampicin (als Enzyminduktor) und Naltrexon in der Behandlung des cholestatischen Pruritus in der Regel wirksam. Auch Cholestyramin kann eingesetzt werden, spielt aber aufgrund der komplizierten Verabreichungsregeln und des unangenehmen Geschmacks in der Pädiatrie eine eher untergeordnete Rolle, Phenobarbital wird aufgrund des Nebenwirkungsprofils ebenfalls nur noch selten eingesetzt.
Screeningmethoden
Da Eltern entfärbten Stuhlgang nicht unbedingt als Warnzeichen erkennen (können), ist die Stuhlfarbenkarte ein einfaches, kostengünstiges und effektives Instrument, um eine möglichst frühzeitige Diagnose einer neonatalen Cholestase stellen zu können. Diese ist in anderen Ländern bereits seit Langem in Gebrauch, sie wurde zum Beispiel in Japan bereits in den 1990er-Jahren eingeführt. In Niedersachsen sind im Rahmen eines Pilotprojektes durch die Medizinische Hochschule Hannover und die Techniker Krankenkasse seit Ende 2016 mehr als 200.000 Stuhlfarbenkarten an alle Geburtskliniken verschickt worden, die in die Vorsorgehefte eingelegt werden (Abb. 2) – eine bundesweite Ausweitung ist geplant [12].
- Im Rahmen der U3 sollte die Stuhlfarbe anhand einer Farbenkarte von den Eltern benannt, besser noch durch den Kinderarzt selbst begutachtet werden.
- Jeder Icterus prolongatus sollte nach 2 Wochen (ungestilltes Neugeborenes) bzw. nach 3 Wochen (gestilltes Neugeborenes) durch eine Bilirubindifferenzierung weiter abgeklärt werden.
- Gutes Gedeihen und ein klinisch guter Zustand bei einem ikterischen Kind schließen eine neonatale Cholestase (insbesondere eine Gallengangatresie) nicht aus.
- Die mangelhafte Resorption fettlöslicher Vitamine im Rahmen der Cholestase kann bereits nach kurzer Zeit zu einem deutlichen Vitamin-K-Mangel führen mit der möglichen Folge lebensgefährlicher Blutungen.
- Jede neonatale Cholestase muss zeitnah in einem spezialisierten Zentrum abgeklärt werden; die frühe Diagnosestellung ist bei der Gallengangatresie entscheidend für den Langzeitverlauf nach Hepatoportoenterostomie.
- Wichtigste Differenzialdiagnosen der neonatalen Cholestase sind neben der Gallengangatresie Infektionen, das Alagillesyndrom, die progressive familiäre intrahepatische Cholestase (PFIC), der alpha-1-Antitrypsinmangel, die CF sowie metabolische und endokrinologische Erkrankungen.
- Die weitere stufenweise Diagnostik erfolgt durch eine Sonographie (Gallenblasenrudiment bei der Gallengangatresie), hepatobiliäre Sequenzszintigraphie (ggf. fehlende Darstellung der Gallenausscheidung), ERCP (direkte Gallenwegsdarstellung), Leberbiopsie sowie offene Cholangiographie.
- Die Therapie der Gallengangatresie erfolgt durch eine Hepatoportoenterostomie nach Kasai, die den Galleabfluss in den Darm wiederherstellen soll.
- Nach der Hepatoenterostomie müssen die Kinder weiter in einem pädiatrischen Lebertransplantationszentrum angebunden werden, da die Mehrzahl der Patienten bereits im Kindesalter eine Lebertransplantation benötigt.
- Eine wesentliche Komplikation nach der Kasai-Operation ist die Cholangitis, die sich durch Fieber ohne sonstigen Fokus, Pruritus, ggf. Ikterus und Stuhlentfärbung bemerkbar macht und frühzeitig iv-antibiotisch behandelt werden muss.
| [1] | Päd. Gastroenterologie, Klinik für Kinder- und Jugendmedizin, Universitätsmedizin Göttingen; | |
| [2] | Päd. Gastroenterologie, Hepatologie und Lebertransplantation; Klinik für Pädiatrische Nieren-, Leber- und Stoffwechselerkrankungen, Medizinische Hochschule Hannover |

Dr. Christoph Leiskau
Erschienen in: Kinderärztliche Praxis, 2022; 93 (2) Seite 113-120


